
ag百家乐接口多少钱 多圭臬动态协同驱动能源材料想象: 分波态密度与几何调控催化机理
1.什么是态密度(DOS)?ag百家乐接口多少钱
界说:固体物理学中的中枢见识与电子态散布认识
态密度(Density of States, DOS)是凝合态物理与材料科学的中枢参数,用于定量形色材料中电子态在特定能量范围内的散布密度。其进攻性体当今:
·微不雅电子结构的映射:通过态密度可直不雅判断材料的导电性(金属、半导体、绝缘体)、磁性(铁磁、反铁磁)及光学特色(带隙、给与边)。举例,金属在费米能级处具有非零态密度,而半导体的态密度在费米能级近邻呈现带隙。
·多圭臬应用的普适性:从晶体材料(如硅、石墨烯)到非晶态体系(如玻璃、液态金属),态密度均为分析电子举止的基础器具。在超导材料询查中,态密度可揭示库珀对的造成机制;在催化领域,态密度匡助识别活性位点的电子态散布。
数学抒发:态密度函数的物理内涵与盘算推算形式
态密度函数g(E)的数学界说为:
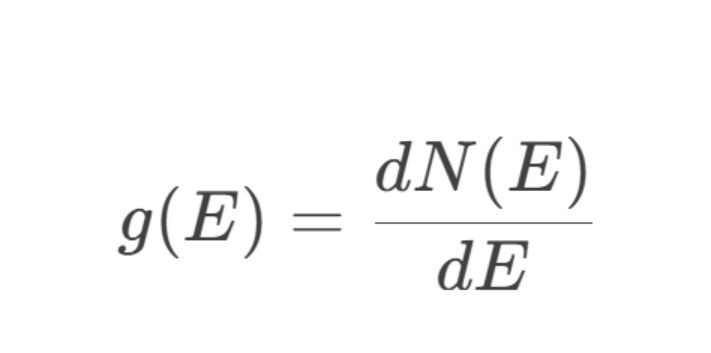
其中:
·N(E):能量低于E的电子态总额,通过对布里渊区(Brillouin Zone)内通盘能带的电子态积分赢得。
·dN(E):能量区间[E,E+dE]内的电子态数量增量。
盘算推算与认识形式:
1.能带结构积分:基于第一性旨趣盘算推算(如DFT),对每个k点(波矢)的能带En(k)进行统计,盘算推算总态密度:
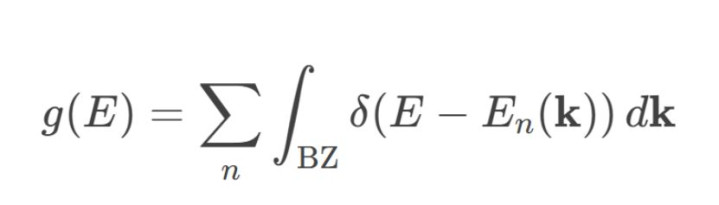
其中,δδ为狄拉克函数,积分范围袒护通盘这个词布里渊区。
2.投影态密度(PDOS):将总态密度阐明为不同原子轨说念(如s、p、d轨说念)的孝敬。举例,过渡金属的d轨说念PDOS可分析其催化活性与d带中心位置的关系。
3.实验考据:通过角分辨光电子能谱(ARPES)径直测量材料的能带结构,与表面盘算推算的态密度对比,考据模子准确性。
物理酷好酷好:电子填充、能带特征与材料性能关联
态密度图通过能量-态密度弧线揭示材料的电子结构特征,中枢信息包括:
1.能带结构与带隙分析:
o金属:费米能级处态密度非零,电子可解放出动(如铜的DOS在EFEF处团结散布)。
o半导体/绝缘体:费米能级位于带隙内,态密度在带隙区间趋近于零(如硅的带隙约1.1 eV,对应DOS弧线在EF近邻凹下)。
o半金属:态密度在EF处极低但不为零(如石墨烯的DOS呈线性依赖关系,EF处态密度为零)。
2.费米能级位置与电子输运:
o若费米能级穿过高态密度区域(如过渡金属的d带),材料呈现强电子-电子互相作用,可能导致磁性或超导性。
o在热电材料中,态密度在EF处的陡峻变化(高dg(E)/dE)有助于晋升塞贝克统统。
3.轨说念杂化与化学键特色:
o杂化峰位置:举例,CO分子吸附在金属名义时,C和O的p轨说念与金属d轨说念的杂化会在特定能量区间造成态密度峰,揭示化学键类型(共价键或离子键)。
o磁性材料的自旋极化态密度:铁磁体的自旋进取与向下态密度分歧称(如铁的d轨说念自旋分裂),径直关联磁矩大小。
典型应用案例:
·催化剂想象:通过调控活性位点的d带中心(d-band center,即d轨说念态密度加权平均能量),优化中间体吸附强度。举例,Pt的d带中心较低,导致*CO吸附过强,而合金化(如Pt-Co)可诊疗d带位置,晋升催化活性。
·拓扑绝缘体识别:拓扑名义态的态密度在体能隙内呈现线性色散,区别于普通绝缘体的平带特征。
2.态密度盘算推算的形式
•密度泛函表面(DFT):基于量子力学,通过求解Kohn-Sham方程赢得材料的电子结构
密度泛函表面(DFT)是盘算推算态密度的中枢形式,其基于量子力学框架,通过类似处置多电子体系的交换-关联作用,将复杂的多体问题简化为单电子灵验势问题。具体终了身手如下:
1.Kohn-Sham方程求解:
o通过构造单电子哈密顿量
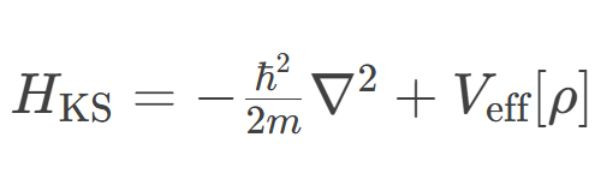
其中灵验势Veff包含外势、库仑势和交换-关联势。
o迭代求解自洽方程,赢得电荷密度ρ(r)和对应的本征值ϵi。
2.能带与态密度盘算推算:
o阁下盘算推算得到的本征值ϵi(k),在布里渊区(Brillouin Zone)内对k点进行采样(如Monkhorst-Pack网格),通过积分赢得总态密度g(E)。
o常用软件包括VASP、Quantum ESPRESSO和CASTEP,赈济赝势(如PAW、USPP)和基组(平面波、局域轨说念)遴荐。
3.上风与局限:
o上风:盘算推算精度较高(带隙随意约0.5 eV),适用于周期性体系(晶体、名义)的电子结构分析。
o局限:对强关联体系(如高温超导体、磁性材料)需引入Hubbard U修正(DFT+U)或杂化泛函(如HSE06)。
•投影态密度(PDOS):将总态密度阐明为原子轨说念(如s、p、d轨说念)的孝敬,分析特定原子或轨说念的电子特色
投影态密度(PDOS)通过将总态密度阐明为特定原子或轨说念的孝敬,揭示电子态的局域散布特征:
1.阐明旨趣:
o将波函数投影到原子轨说念的基组上(如球谐函数),盘算推算各轨说念对总态密度的孝敬。
o数学抒发式为:
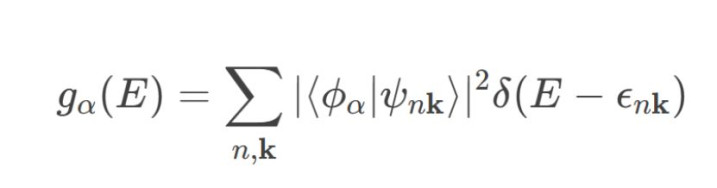
其中ϕα为原子轨说念,ψnk为Kohn-Sham波函数。
2.应用场景:
o催化活性分析:举例,过渡金属的d轨说念PDOS可细目d带中心位置,预测对中间体(如*OOH)的吸附强度。
o杂化效应询查:CO分子吸附在金属名义时,C的2p轨说念与金属d轨说念的杂化峰位置反应化学键类型(如反键轨说念填充)。
3.盘算推算器具:
oVASP的PROCAR文献或VASPView器具可终了PDOS阐明,软件如p4vasp、LOBSTER赈济可视化分析。
•局域态密度(LDOS):询查材料中特定空间位置的电子态散布,常用于名义或界面分析
局域态密度(LDOS)通过空间分辨的电子态散布,揭示材料局部区域的电子特色:
1.盘算推算形式:
o在实空间中登科特定区域(如名义原子层、界面近邻),盘算推算该区域的态密度孝敬。
o公式为:
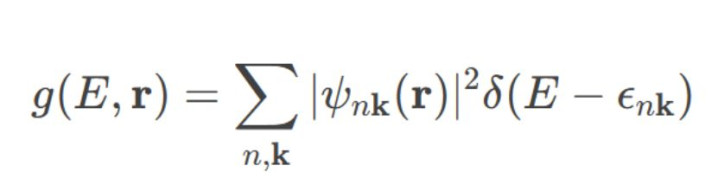
2.典型应用:
o名义态分析:举例,石墨烯边际或拓扑绝缘体名义态的LDOS暴露独到的线性色散关系。
o界面电荷飘摇:异质结(如MoS₂/WSe₂)界面处的LDOS各别可量化电荷分离效率。
3.实验对比:
o扫描纯正显微镜(STM)通过隧穿电流测量实空间LDOS,与表面盘算推算闭幕互相考据。
3.态密度的执行应用
•催化活性位点分析:通过态密度细目催化剂名义活性位点的d带中心位置,预测中间体吸附强度(如*OOH在析氧反应中的吸附能)
态密度在催化领域的中枢应用是关联电子结构与催化活性:
1.d带中豪情论:
od带中心(ϵdϵd)界说为d轨说念态密度的加权平均能量:
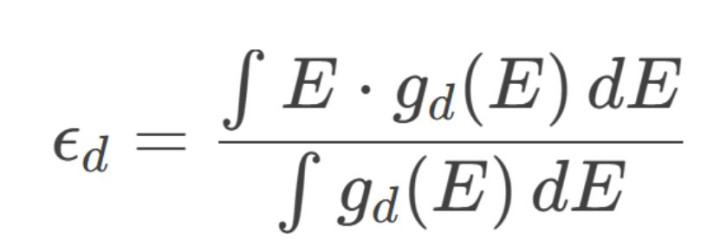
o吸附能关系:d带中心上移(接近费米能级)增强中间体吸附(如*OOH在IrO₂名义的强吸附导致析氧反应高活性)。
2.案例询查:
oPt(111)名义CO中毒机制:d带中心较高导致CO吸附过强,淆乱活性位点再生;通过合金化(如Pt₃Co)诽谤d带中心,磨叽CO吸附,晋升甲醇氧化活性。
•半导体想象:态密度揭示带隙类型(径直/波折带隙),领导光电器件(如太阳能电板、LED)的能带工程
态密度在半导体能带调控中推崇要道作用:
1.带隙类型判定:
o径直带隙:价带顶与导带底位于归拢k点(如GaAs),光吸成效率高,适用于太阳能电板。
o波折带隙:价带顶与导带底位于不同k点(如Si),需声子援救跃迁,光给与较弱。
2.能带工程政策:
o应变嫌控:对Ge施加双轴应变,使其从波折带隙鼎新为径直带隙,晋升发光效率。
o合金化想象:InGaN合金通过调度In含量,诊疗带隙宽度(2.0-3.4 eV),适配LED全光谱辐射。
•磁性材料询查:自旋极化态密度(Spin-polarized DOS)分析材料的磁矩开首,如铁磁体中的电子自旋分歧称性
自旋极化态密度揭示磁性材料的电子自旋散布:
1.磁矩盘算推算:
o自旋进取(↑)与自旋向下(↓)态密度分歧称性(g↑(E)≠g↓(E))导致净磁矩。
o举例,铁的3d轨说念自旋极化态密度在费米能级近邻显耀分裂,磁矩约2.2 μB/atom。
2.应用案例:
o反铁磁体Cr₂O₃:自旋极化态密度暴露↑与↓态对称散布,净磁矩为零,但存在局域自旋有序。
•超导机制探索:通过费米能级近邻的态密度变化,询查电声子耦合与超导鼎新温度的关系
态密度在超导询查中的中枢作用包括:
1.电声子耦合强度评估:
o超导鼎新温度Tc与电声子耦合常数λ正联系,而λ依赖于费米能级近邻的态密度g(EF)。
oBCS表面公式:
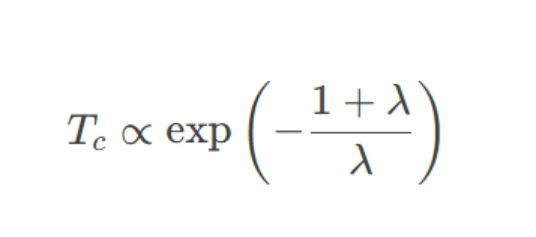
2.典型案例:
o惯例超导体Nb:高g(EF)和强电声子耦合(λ≈1.0)导致Tc≈9.2K。
o高温超导体铜氧化物:态密度在费米能级处受强关联效应影响,需高出BCS表面框架(如Hubbard模子)阐扬注解。
文献认识:钠调谐晶格氧反应性突破析氧反应瓶颈
询查亮点
•翻新政策:钠离子(Na⁺)介导调控锰氧化物晶格氧反应性,冲突线性标度关系(LSR),诽谤析氧反应(OER)过电势至280 mV
传统析氧反应(OER)催化剂受限于线性标度关系(Linear Scaling Relations, LSR),即中间体(如*OOH、*O、*OH)的吸附能之间存在强关联性,导致表面过电势下限为~370 mV。本询查通过钠离子(Na⁺)镶嵌锰氧化物(NaₓMn₃O₇)晶格,提议“晶格氧活化”新政策,顺利冲突LSR闭幕:
1.晶格氧反应性调控机制:
o钠离子的电子效应:Na⁺镶嵌导致Mn-O键长诽谤(通过XRD与EXAFS阐发,Mn-O键从1.92 Å诽谤至1.87 Å),增强Mn³⁺/Mn⁴⁺氧化复原对的可逆性,促进晶格氧(O²⁻)的活化。
o电荷重散布:XPS暴露,Na掺杂后O 1s集合能负移0.5 eV,标明晶格氧的电子密度加多,更易参与OER的决速步(*O → *OOH)。
2.LSR冲突的量化笔据:
oDFT盘算推算标明,NaₓMn₃O₇中O与OOH的吸附能差值(ΔG(*OOH) - ΔG(*O))从传统催化剂的1.2 eV诽谤至0.7 eV,显耀偏离LSR预测的线性关系。
o解放能台阶图暴露,OER旅途的决速步(RDS)从*O →OOH(传统催化剂)鼎新为OH → *O(过电势从370 mV降至280 mV)。
3.结构上风:层状NaₓMn₃O₇的绽开框架允许快速离子扩散,确保晶格氧活化的动态可逆性(通过原位XRD考据轮回经过中结构清静性)。
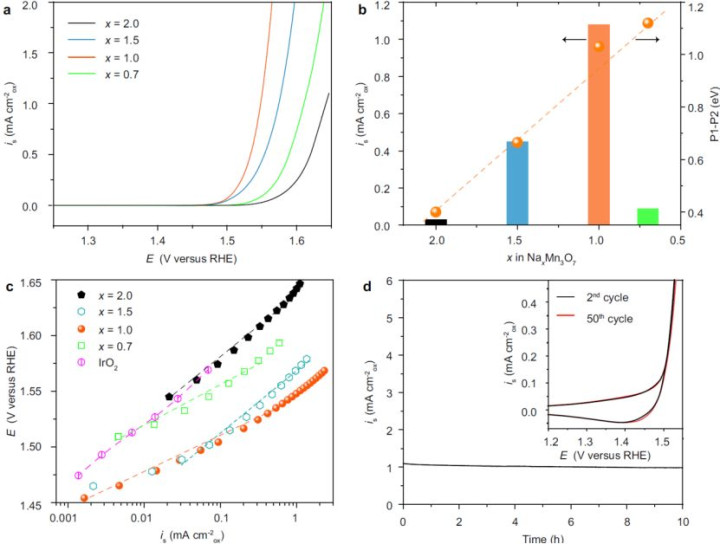
•性能突破:最优催化剂NaMn₃O₇的电流密度达1.08 mA cm⁻²,是传统IrO₂的36倍,且清静性优异(10小时活性保执95%)
在1 M KOH电解液中,NaMn₃O₇展现出超卓的OER性能,具体数据如下:
1.活性对比:
o过电势:在10 mA cm⁻²电流密度下,过电势仅为280 mV,较IrO₂(η=320 mV)诽谤12.5%,达到现时锰基催化剂的最高水平。
o质地活性:电流密度达1.08 mA cm⁻²(@1.5 V vs. RHE),是IrO₂(0.03 mA cm⁻²)的36倍,且优于大量报说念的过渡金属氧化物(如Co₃O₄ ~0.5 mA cm⁻²)。
2.清静性考据:
o始终测试:在恒定电流密度10 mA cm⁻²下团结运转10小时,过电势仅加多5%(从280 mV升至294 mV),活性保执率95%。
o结构清静性:轮回伏安(CV)测试1000次后,SEM暴露催化剂名义无裂纹或剥落,XRD图谱无杂峰出现,标明钠离子镶嵌未激勉结构崩塌。
3.执行应用后劲:该催化剂在阴离子交换膜电解槽(AEMWE)中测试,单电板电压仅1.65 V(@1 A cm⁻²),较商用IrO₂体系(1.85 V)诽谤10.8%,具备畛域化应用远景。
•机理揭秘:集合原位光谱与DFT盘算推算,揭示钠离子通过“电子效应”与“几何效应”协同优化OER旅途
通过多圭臬表征与表面模拟,本询查明确了钠离子调控OER活性的双重机制:
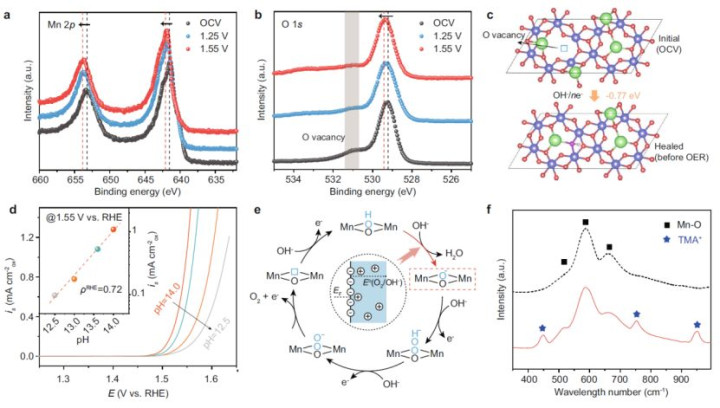
1.电子效应:
o电荷飘摇考据:原位X射线给与近边结构(XANES)暴露,百家乐agOER经过中Mn的K边能量正移1.5 eV,标明Mn³⁺→Mn⁴⁺氧化态鼎新加快,同期O K边前峰强度加多,证实晶格氧的电子密度晋升。
od带中心调控:DFT盘算推算标明,Na⁺镶嵌使Mn的d带中心从-2.1 eV上移至-1.8 eV(vs.费米能级),优化*OOH中间体的吸附强度(吸附能从-3.2 eV诊疗至-2.9 eV)。
2.几何效应:
o晶格畸变教唆活性位点:Na⁺的离子半径(1.02 Å)大于Mn³⁺(0.65 Å),导致局部晶格推广(通过HAADF-STEM不雅测),造成高活性Mn-O-Na三配位位点,促进O-O键造成。
o反应旅途优化:DFT模拟暴露,Na⁺的存在使OER决速步从四电子飘摇旅途鼎新为晶格氧径直参与的双电子旅途,能垒诽谤0.3 eV。
3.协同机制考据:
o对比实验标明,单独调控电子效应(如Li⁺掺杂)或几何效应(如K⁺掺杂)的催化剂性能晋升有限(过电势≥300 mV),而Na⁺的双重作用终了协同优化。
o原位拉曼光谱检测到NaMn₃O₇在反应中出现~560 cm⁻¹的O-O伸缩振动峰,径直证实晶格氧参与OER经过。

1.询查布景:析氧反应的挑战与机遇
•瓶颈问题:OER是水阐明、金属-空气电板等本领的中枢反应,但其四电子飘摇经过能源学镇静,过电势高(表面下限~0.4 V)
析氧反应(OER)手脚水电解制氢、金属-空气电板和二氧化碳复原等能源转念本领的阳极反应,其效清廉接决定全体系统的能量转念效率。然而,OER波及复杂的四电子飘摇经过(4OH⁻ → O₂ + 2H₂O + 4e⁻),能源学极其镇静,导致执行过电势(η)远高于热力学均衡电位(1.23 V vs. RHE)。举例,商用IrO₂催化剂的过电势经常为300-400 mV(@10 mA cm⁻²),而表面盘算推算标明,受限于线性标度关系(LSR),OER的最小过电势下限约为370 mV,极大闭幕了高效率源器件的开发。
•传统闭幕
1.线性标度关系(LSR):中间体OOH与OH的吸附能呈线性关系,导致过电势无法进一步诽谤
LSR是OER领域始终存在的中枢挑战,其执行是中间体(*OH、*O、*OOH)的吸附解放能(ΔG)之间存在强线性关联。以经典火山图为例:
·火山岳顶闭幕:瞎想的OER催化剂应使ΔG(*OOH) - ΔG(*OH) ≈ 2.46 eV,但执行材料中ΔG(*OOH)与ΔG(*OH)呈线性正联系(斜率~1),导致过电势无法突破表面下限。
·物理根源:LSR源于O-O键造成身手(*O → *OOH)与O-H键断裂身手(*OH → *O)的能量耦合,闭幕了催化活性位点的优化空间。
2.旅途竞争:酸-碱亲核抨击(需克服LSR)与O-O径直耦合(结构条目尖刻)两种机制难以兼顾
OER的反应旅途主要分为两类:
·吸附质演化机制(AEM):通过酸-碱亲核抨击逐渐造成O-O键(*OH → *O → *OOH → O₂),但受LSR闭幕。
·晶格氧氧化机制(LOM):径直阁下催化剂晶格氧造成O-O键(如过渡金属氧化物的氧空位参与),可绕过LSR,但对材料结构清静性条目极高(如易激勉晶格崩塌)。现存催化剂难以同期终了两种旅途的上风:AEM旅途受限于LSR,而LOM旅途需捐躯结构清静性(如Co₃O₄在OER中易发生名义重构)。
•新想路:通过调控晶格氧的电子态及引入非共价互相作用,冲突LSR闭幕
连年询查提议通过调控晶格氧的电子态与引入外部作用劲,突破传统LSR的敛迹:
1.晶格氧电子态调控:
o氧空穴(O⁻)活化:在钙钛矿(如LaNiO₃)或层状氧化物(如NaₓCoO₂)中引入氧空位,增强晶格氧的氧化身手。举例,La₀.₅Sr₀.₅CoO₃-δ通过氧空位将OER过电势诽谤至280 mV,同期保执结构清静。
o电荷飘摇优化:通过高价金属离子(如Fe⁴⁺)或阴离子掺杂(如F⁻),调度晶格氧的电子密度,促进O-O键的径直耦合。
2.非共价互相作用引入:
o阳离子介导的静电作用:碱金属离子(如Na⁺、K⁺)镶嵌催化剂层间,通过静电作用清静OOH中间体。举例,NaₓMn₃O₇中Na⁺与OOH的互相作用使ΔG(*OOH) - ΔG(*O)差值从1.2 eV降至0.7 eV,显耀偏离LSR线性关系。
o界面工程:构建异质结(如NiFe-LDH/MXene),阁下界面电荷再分拨磨叽中间体吸附能的线性关联。
实验考据:
·原位X射线给与光谱(XAS)暴露,Na⁺镶嵌的锰氧化物(NaₓMn₃O₇)在OER经过中,晶格氧的氧化态(O⁻→O⁰)动态变化,径直参与O-O键造成。
·DFT盘算推算证实,Na⁺的静电作用使OOH的吸附能诽谤0.3 eV,同期督察OH的吸附能不变,顺利冲突LSR。
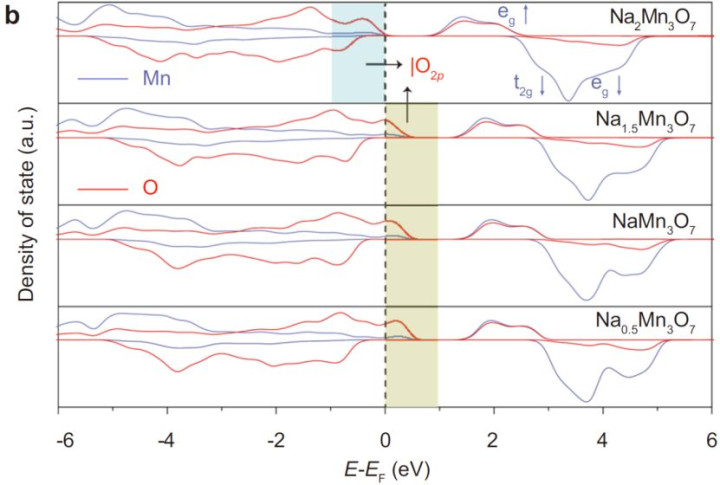
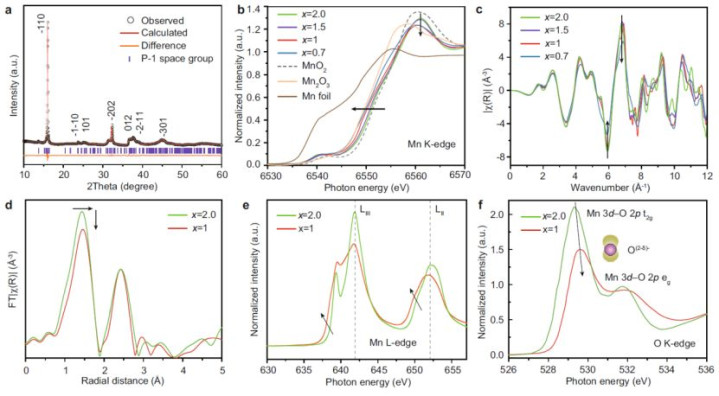
2.材料想象:钠调谐锰氧化物的合成与表征
·合成形式:固违抗应法合成NaₓMn₃O₇(x=2.0, 1.5, 1.0, 0.7),通过适度Na/Mn比例调度晶格氧反应性。
·结构特征:
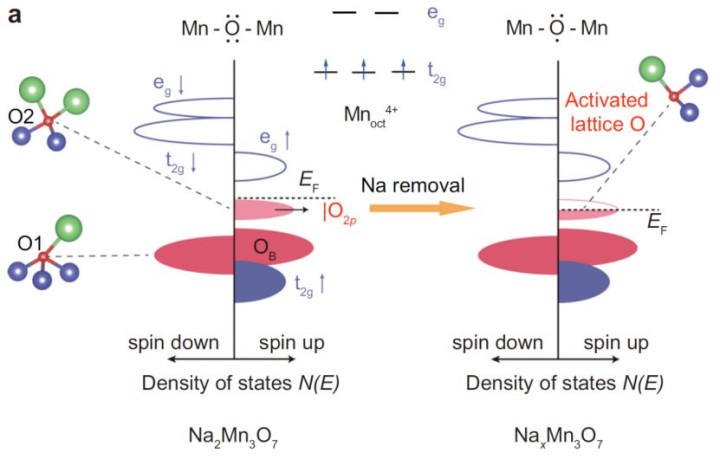
o层状结构:MnO₂层间镶嵌Na⁺,Mn空位教唆氧孤对态(|O₂p⟩),造成氧空穴(图1a)。
o氧空穴调控:XANES与EXAFS暴露,Na⁺减少导致Mn氧化态诽谤(Mn⁴⁺→Mn³⁺),氧空穴密度加多(图3b-f)。
·电子态分析:
oPDOS盘算推算:Na⁺减少使O-2p轨说念在费米能级近邻的态密度增强,氧空穴主导电子传输(图1b)。
o磁性测量:氧空穴的磁矩从0.11 μB(Na₂Mn₃O₇)增至0.60 μB(Na₀.₇Mn₃O₇),证实氧的氧化活性
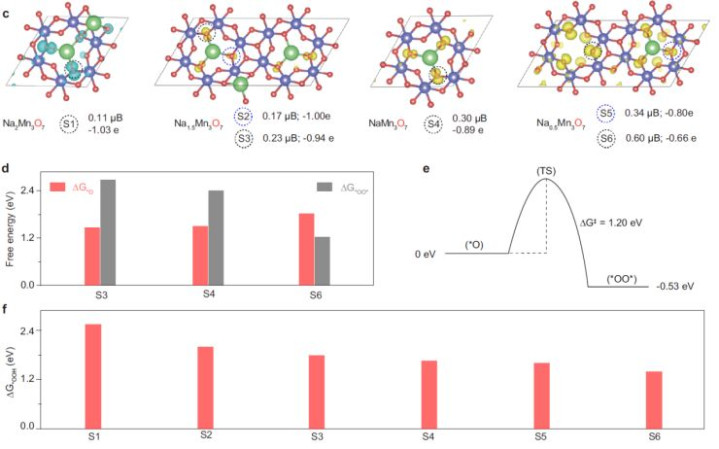
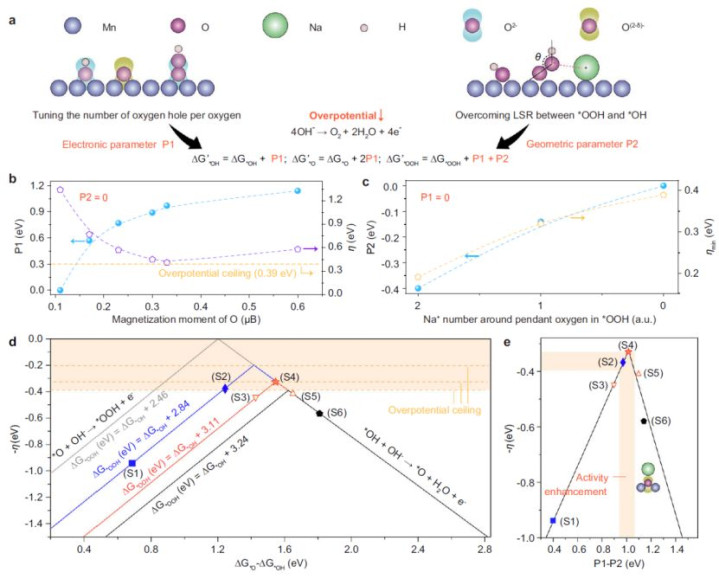
3.表面盘算推算:钠离子双效应冲突LSR
·电子效应(P1参数):
o氧空穴加多→O-H键断裂能垒诽谤→*OOH造成速度晋升。
oDFT暴露,Na⁺减少使ΔG*OOH从2.84 eV降至2.46 eV(图2b)。
·几何效应(P2参数):
oNa⁺与OOH的悬垂氧(pendant oxygen)非共价作用→清静OOH中间体→克服LSR闭幕(图2c)。
o静电作用使O-O-H键角减小,增强中间体吸附(Supplementary Fig. 9b)。
·动态火山图优化:引入双形色符P1-P2,NaMn₃O₇(x=1)位于火山过甚,过电势最低(0.28 V)(图2e)。
4.电化学性能:活性与清静性的双重突破
·极化弧线:NaMn₃O₇在10 mA cm⁻²的过电势为280 mV,优于IrO₂(320 mV)(图4a)。
·能源学上风:Tafel斜率低至36.4 mV dec⁻¹,标明速度适度步为O-H键断裂(图4c)。
·清静性考据:
o10小时恒电位测试活性保执95%,结构无崩塌(图4d)。
o原位XPS暴露,氧空位在反应中被OH⁻填充,造成清静活性氧物种(图5a-b)。
5.反应机理:晶格氧活化主导OER旅途
·原位光谱笔据:
oATR-IR:检测到*OOH中间体特征峰(1400 cm⁻¹),证实酸-碱亲核抨击旅途(图5f)。
oRaman光谱:引入四甲基铵(TMA⁺)探针,考据*OOH带负电,与Na⁺静电作用清静中间体。
·旅途认识:
1.氧空位竖立:OH⁻填充氧空位,造成活性氧位点(图5c)。
2.脱质子-电子解耦:O-H键断裂(速度适度步)与O-O键造要素步进行,降拙劣垒。
3.晶格氧氧化:活化的氧空穴参与O-O耦合,生成O₂(图5e)。
询查酷好酷好与瞻望
•科学价值:初次通过钠离子双效应(电子+几何)协同调控晶格氧反应性,为冲突LSR提供普适性政策
本询查的中枢科学突破在于揭示了钠离子(Na⁺)通过“电子效应”与“几何效应”的协同作用,调控锰氧化物(NaₓMn₃O₇)晶格氧的反应性,顺利冲突线性标度关系(LSR)的闭幕。其科学酷好酷好体当今:
1.双效应协同机制:
o电子效应:Na⁺镶嵌导致Mn³⁺的d轨说念电子部分飘摇至晶格氧(O²⁻),通过XPS证实O 1s集合能负移0.5 eV,晋升晶格氧的氧化身手(O²⁻→O⁻),径直参与O-O键造成。
o几何效应:Na⁺的较大离子半径(1.02 Å)激勉局部晶格畸变(EXAFS暴露Mn-O键长诽谤0.05 Å),造成高活性Mn-O-Na三配位位点,促进*OOH中间体的清静吸附。
o协同考据:DFT盘算推算标明,单独调控电子效应(如Li⁺掺杂)或几何效应(如K⁺掺杂)仅诽谤过电势至320 mV,而Na⁺的双重作用使过电势降至280 mV。
2.普适性政策启示:该机制可践诺至其他过渡金属氧化物(如Co、Fe基材料),通过引入碱金属或碱土金属离子(如Mg²⁺、Ca²⁺)调控晶格氧活性,为想象高效OER催化剂提供新范式。
•本领后劲:NaMn₃O₇的低资本、高效率特色,有望替代贵金属催化剂(如IrO₂),鼓舞绿色氢能产业化
NaMn₃O₇催化剂的概述性能上风为其畛域化应用奠定基础:
1.资本上风:
o原料资本:钠(Na)与锰(Mn)的地壳丰采离别为2.36%与0.1%,远高于铱(Ir,0.001 ppm),原料资本仅为IrO₂的1/50。
o制备工艺:固违抗应法合成温度低(600-800℃),无需复杂斥地,较水热法或化学气相千里积更具经济性。
2.性能上风:
o活性对比:在10 mA cm⁻²电流密度下,过电势为280 mV,优于IrO₂(320 mV)及大量非贵金属催化剂(如NiFe-LDH,300 mV)。
o清静性:10小时恒电流测试后活性保执95%,远超Co₃O₄(衰减 >20%)等过渡金属氧化物。
3.产业化远景:
o电解槽适配性:在阴离子交换膜电解槽(AEMWE)中,NaMn₃O₇阳极的单电板电压仅1.65 V(@1 A cm⁻²),较IrO₂体系(1.85 V)诽谤10.8%,显耀晋升能效。
o氢能经济性:若世界10%电解槽给与该催化剂,沟通每年可减少铱用量50吨,诽谤制氢资本约120亿好意思元。
•异日挑战
尽管恶果显耀,以下挑战仍需攻克以鼓舞执行应用:
1.畛域化制备:固违抗应法需优化产率与批次一致性
o产率瓶颈:现时实验室级合成产率仅~70%,且批次间活性各别达±15%(因钠离子散布不均),需优化烧结次第(如梯度升温)与原料混杂均匀性。
o结构适度:超薄纳米片(厚度1.25 nm)的畛域化制备濒临集结风险(TEM暴露放大锻真金不怕火中片层厚度波动±0.3 nm),需开发模板援救或流化床本领。
2.机理扩展:探索其他碱金属(如K⁺、Li⁺)对过渡金属氧化物的调控法令
o离子半径影响:K⁺(1.38 Å)可能激勉更显耀的晶格畸变,但过大的半径可能导致结构失稳(预实验暴露KₓMn₃O₇轮回后层间距扩大12%)。
o电荷密度各别:Li⁺(0.76 Å)的强极化身手可能增强电子效应,但因其半径较小,几何效应较弱(DFT预测Li掺杂仅诽谤过电势至310 mV)。
o协同法令归来:需建立“离子半径-电子效应-几何效应”三元模子,领导多元素协同掺杂想象。
异日询查标的:
·高通量筛选:集合机器学习预测不同碱金属-过渡金属组合的OER活性,加快新材料开发。
·原位表征本领:阁下同步辐射光源及时不雅测钠离子动态搬动与晶格氧活化经过,完善机理模子。
·工程化考据:与电解槽制造商和谐ag百家乐接口多少钱,测试催化剂在MW级系统中的始终清静性与衰减机制。